






Marfan sendromu nedir? Bu sorunun cevabını genetik bilimini aralayarak tanımlanabilir: Marfan sendromu, FBN1 genindeki mutasyonlar nedeniyle ortaya çıkan ve otozomal dominant kalıtımla nesilden nesle aktarılan bir hastalıktır. Bu nadir hastalık bağ dokusunun yapısını bozarak vücudun birçok sistemini etkiliyor. Kalpten gözlere, iskelet sisteminden kan damarlarına kadar uzanan bu etkiler Marfan sendromunu hem gizemli hem de hayati bir durum haline getiriyor. Peki marfan sendromu belirtileri neler ve marfan sendrom tedavisi nasıl gerçekleşiyor?
Marfan sendromunun tarihi ve gelişimi
Marfan sendromu nedir sorusunu cevaplamak için önce bu hastalığın tarihteki orijinine inmek en doğrusu. 1896’da Fransız doktor Antoine Marfan; beş yaşındaki bir kız çocuğunda uzun kollar, bacaklar ve ince bir vücut yapısı gibi belirtileri fark etti. Bu gözlem, bugün Marfan Sendromu olarak bilinen nadir bir hastalığın keşfine yol açtı.
Marfan sendromu, yaklaşık her 5.000 kişiden birinde görülen ve vücuttaki bağ dokusunu etkileyen genetik bir hastalıktır. FBN1 genindeki bir mutasyon, vücudun yapısal desteğini sağlayan fibrillin proteininin işlevini bozuyor. Bu da iskelet, kalp-damar sistemi ve gözler gibi birçok yapıyı etkileyebiliyor.
Marfan sendromunun keşfi, bilim insanlarının bağ dokusunun sırlarını çözme çabalarıyla şekillendi. 1980’lerde fibrillin proteininin keşfi, hastalığın moleküler temelini aydınlattı. Bu protein, vücuttaki elastik liflerin yapısını korumak ve hücreler arası iletişimi düzenlemek için kritik bir rol oynuyor. 1991’de FBN1 geninin keşfi ise hastalığın kalıtsal yönünü netleştirdi.
Marfan sendromu, kişiden kişiye farklı şekillerde ortaya çıkabiliyor. İşte tam olarak bu noktada Marfan sendromu nedir ifadesi farklı şekillerde cevap bulabiliyor. Bazı hastalarda uzun boy ve ince yapı gibi hafif belirtiler görülürken diğerlerinde kalp-damar sorunları veya göz problemleri gibi ciddi komplikasyonlar gelişebiliyor. Erken teşhis ve doğru tedavi, özellikle kalp-damar sistemini koruyarak yaşam süresini uzatabiliyor. Örneğin, birinci kalp krizinden sonra ikinci kalp krizini önleyici beta blocker gibi ilaçlar, aort damarının genişlemesini yavaşlatarak hayati riskleri azaltıcı etki gösterebiliyor.

FBN1 genindeki mutasyonlar
Marfan sendromunun genetik temelinde, FBN1 genindeki mutasyonlar yer alır. Bu gen, vücudumuzdaki bağ dokusunun esnekliğini ve dayanıklılığını sağlayan fibrillin-1 adlı bir proteinin üretiminden sorumludur. Fibrillin-1, adeta bir iskele gibi çalışarak dokuların yapısını destekler. Ancak FBN1 genindeki mutasyonlar, bu proteinin yapısını bozar ve bağ dokusunun zayıflamasına neden olur. Bu da Marfan sendromunun belirtilerinin ortaya çıkmasına yol açar. [2]
| Sendrom adı | Temel belirtiler | Neden olan gen(ler) | Görülme sıklığı | Tedavi | Ortalama yaşam süresi |
|---|---|---|---|---|---|
| Marfan sendromu | Uzun kol/bacaklar, göz merceği kayması, aort genişlemesi/yırtılması | FBN1, TGFBP2 | 1/3.000 – 5.000 | Beta blokerler, cerrahi (aort >50 mm) | Tedaviyle 70 yıl |
| Doğuştan kasılmalı arachnodaktili | Kısa kemikler, parmak deformitesi, eklem sertliği, hafif aort genişlemesi | FBN2 | Bilinmiyor | Fizik tedavi, yıllık kalp kontrolü | Sorunsuz – normal |
| Loeys-Dietz sendromu (Tip I) | Yüksek tansiyon, damar kıvrımlılığı, yarık damak | TGFBP1, TGFBP2 | Bilinmiyor | Beta blokerler, cerrahi (aort >50 mm) | 37 yıl |
| Loeys-Dietz sendromu (Tip II) | Tip I’e benzer, yüz belirtisi yok | TGFBP1, TGFBP2 | Bilinmiyor | Beta blokerler, cerrahi (aort >50 mm) | 37 yıl |
| Ehlers-Danlos sendromu (Damar tipi) | Kolay morarma, ince cilt, damar/organ yırtılması riski | COL3A1 | 1/25.000 (%4’ü) | Acil cerrahi (riskli durumlarda) | Tedavisiz 48 yıl |
| Shprintzen-Goldberg sendromu | Kafatası deformitesi, göz fırlaklığı, zihinsel gerilik | FBN1 | Bilinmiyor | Kalp kontrolü, ortopedik destek | Bilinmiyor |
| Arteriyal Tortuozite sendromu | Damar kıvrımlılığı, uzun yüz, eklem gevşekliği | SLC2A10 | Bilinmiyor | Damar cerrahisi (gerektiğinde) | Bilinmiyor |
Bugüne kadar FBN1 geninde 500’den fazla farklı mutasyon tespit edildi. Farklı belirtiler ve sorunlar haliyle tek bir marfan sendromu nedir sorusuyla da sonuçlanmadı. Bu mutasyonlar, genin farklı bölgelerinde meydana gelebilir ve her biri proteinin yapısını farklı şekillerde etkileyebilir. İlginç olan aynı mutasyona sahip kişilerde bile hastalığın şiddeti ve belirtileri değişebilir. Örneğin bazı kişilerde kalp-damar sorunları ön plandayken diğerlerinde uzun boy ve ince parmaklar gibi iskelet sistemi belirtileri daha belirgin olabilir.

Bu durum, Marfan sendromunun sadece genetik faktörlerle değil, çevresel etkiler ve diğer genlerle de şekillenebileceğini gösteriyor. Örneğin, bazı hastalarda aort damarının genişlemesi gibi ciddi sorunlar gelişirken diğerlerinde göz lensinin yer değiştirmesi gibi daha hafif belirtiler görülebiliyor. Bu çeşitlilik hastalığın her bireyde farklı bir seyir izlemesine neden oluyor. Tam olarak bu noktadaysa Marfan sendromu belirtileri kısmına bir parantez açmamız gerekiyor.
Marfan sendromu belirtileri
Marfan sendromu nedir diye düşünürken ardından belirtilere dayalı açıklama ihtiyacının gerekliliğine geliyoruz. Marfan sendromu, vücudun farklı sistemlerini etkileyerek çeşitli belirtilere yol açar. Bu belirtiler, hastalığın ne kadar karmaşık ve çok yönlü olduğunu gösterir.
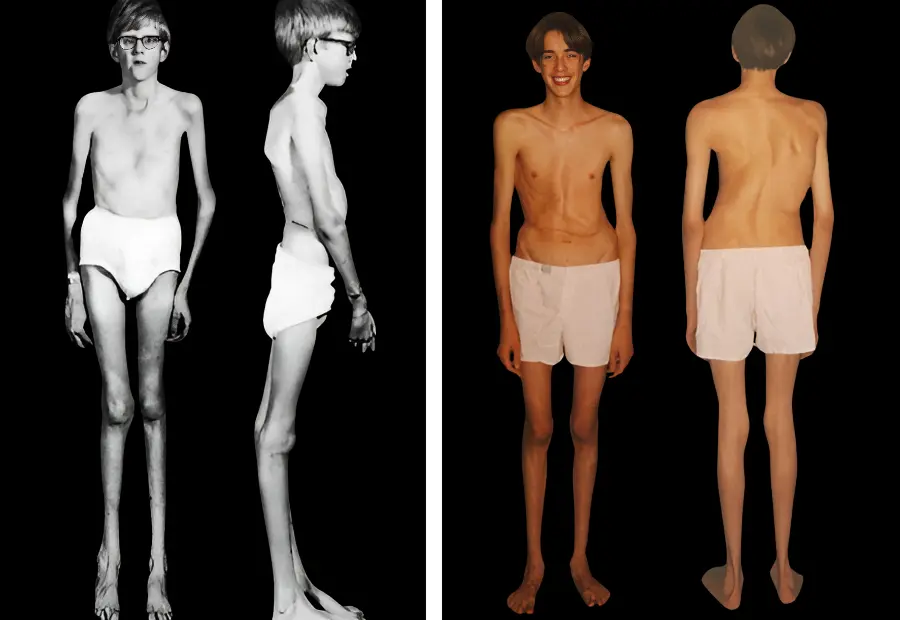
İskelet sistemi, Marfan sendromu belirtileri denince akla ilk gelen sistemlerden birisidir. Hastalar genellikle yaşıtlarına göre daha uzun boylu ve ince yapılıdır. Bu uzunluk; özellikle kollar, bacaklar, el ve ayak parmaklarında belirgindir. Hatta parmaklar o kadar uzun ve ince olabilir ki bu duruma “örümcek parmak” denir. Omurgada eğrilikler (skolyoz) veya kamburluk (kifoz) gibi sorunlar da sıkça görülür. Bu durumlar sadece görünümü değil, hareket kabiliyetini de etkileyebilir. Göğüs kafesinde ise içe çökme veya dışa çıkıntı gibi şekil bozuklukları, solunum fonksiyonlarını bile etkileyebilir.
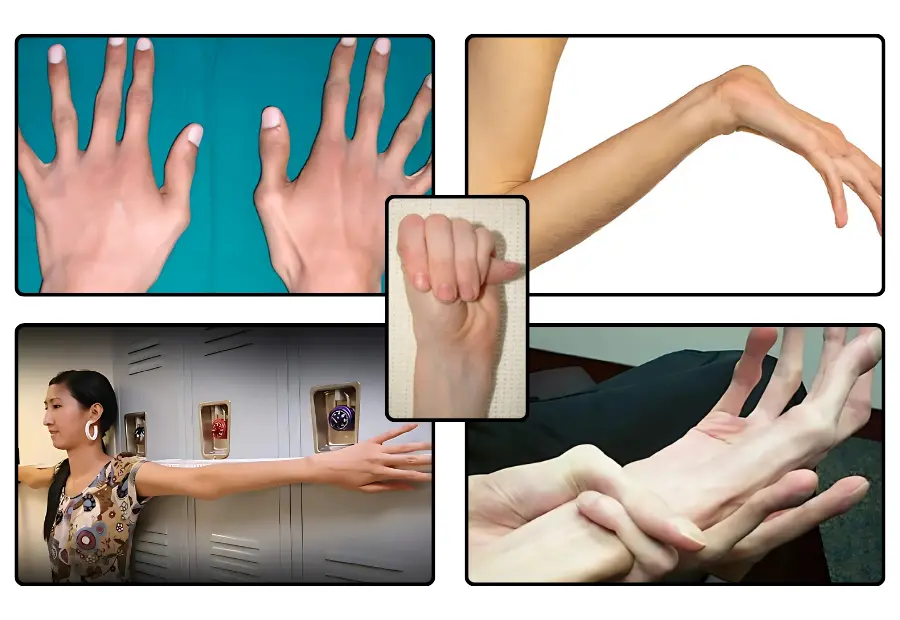


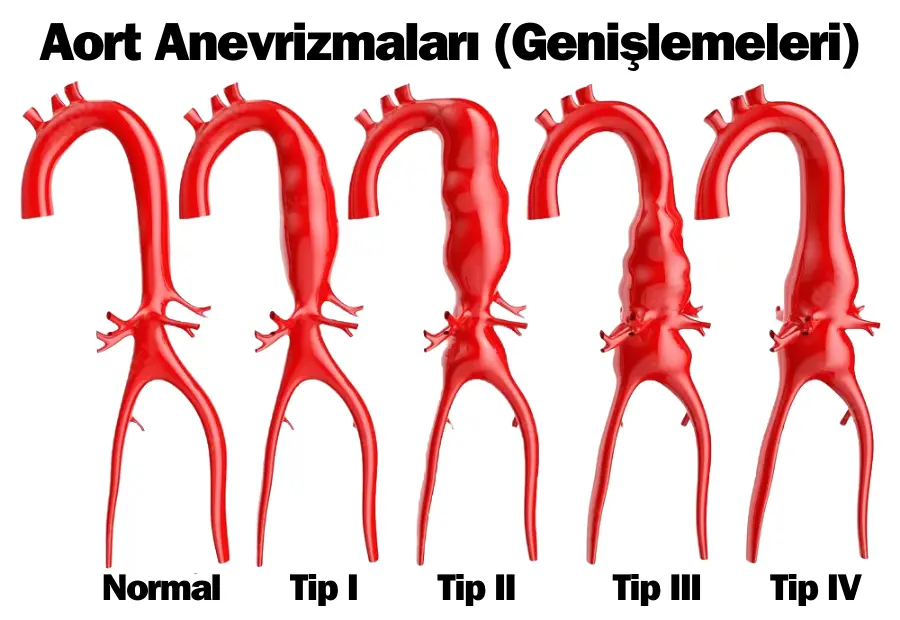
Marfan sendromu belirtileri en ciddi şekilde kalp ve damar sisteminde görülür. Öyle ki marfan sendromu ne demek diye sorunca en yaygın sorunların kardiyovasküler alanla ilgili olduğunu görüyoruz. Aort damarının genişlemesi (aort anevrizması), hastalığın en tehlikeli sonuçlarından biridir. Bu genişleme damarın yırtılma riskini artırır ve bu durum ani ölümlere yol açabilir. Ayrıca kalp kapakçıklarında gevşeme (mitral kapak prolapsusu) gibi sorunlar da sıkça görülür. Bu durum kalbin kan pompalama işlevini bozabilir ve zamanla kalp yetmezliğine neden olabilir. Düzensiz kalp atışları (aritmiler) ise hastaların yaşam kalitesini önemli ölçüde etkileyebilir.

Gözler de Marfan sendromundan etkilenen önemli organlardan biridir. Göz merceğinin yerinden kayması (lens çıkığı), bu sendromun tipik belirtilerinden biridir. Bu durum bulanık görme, çift görme veya ışık hassasiyetine neden olabilir. Ayrıca Marfan sendromlu bireylerde miyopi (uzağı net görememe) sıkça görülür. Retinanın yerinden ayrılması (retina dekolmanı) gibi ciddi bir durum ise acil tedavi gerektirir ve görme kaybına yol açabilir. Glokom (göz içi basıncının artması) ve katarakt (lens bulanıklığı) gibi göz problemleri de daha erken yaşlarda ortaya çıkabilir.

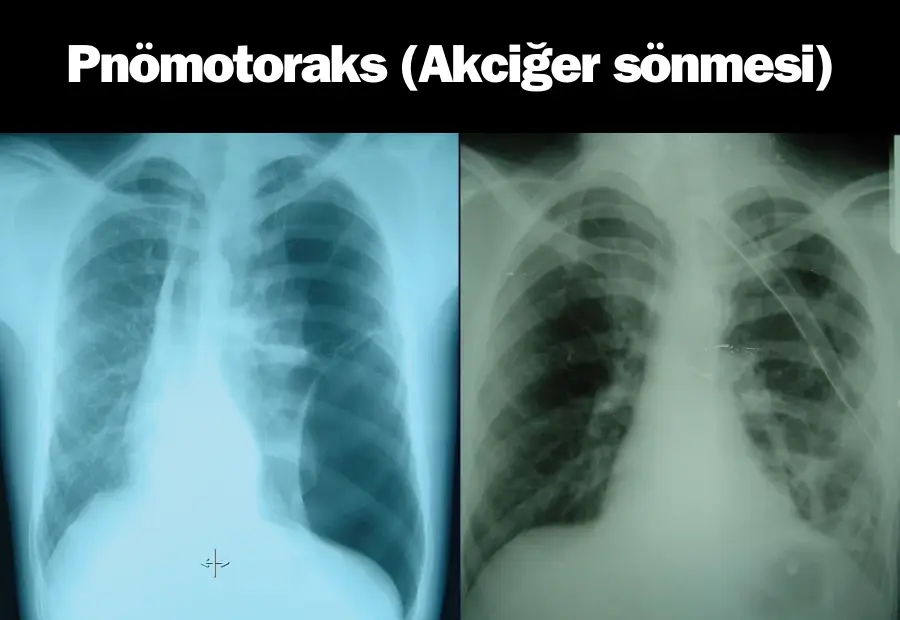
Marfan sendromu belirtileri kendini akciğerde de kendini gösterebilir. Bağ dokusunun zayıflığı, akciğer çökmesi (spontan pnömotoraks) gibi nadir ancak ciddi bir duruma yol açabilir. Bu durum, ani göğüs ağrısı ve nefes darlığı ile kendini gösterir ve acil tıbbi müdahale gerektirir. Ayrıca göğüs kafesi deformiteleri nedeniyle akciğerlerin genişleme kapasitesi kısıtlanabilir, bu da nefes darlığına ve egzersiz yapmada zorluklara neden olabilir. Uyku apnesi gibi solunum problemleri de hastaların yaşam kalitesini olumsuz etkileyebilir.
Marfan sendromu tanı kriterleri
“Marfan sendromu nedir ve devamında nasıl teşhis edilir”i cevaplamak, bir bulmacayı çözmeye benzer. Marfan sendromu belirtileri o kadar çeşitlidir ki, doktorların doğru tanıya ulaşmak için bir dizi ipucunu bir araya getirmesi gerekir. Bu süreçte, 1996’da revize edilen “Ghent Kriterleri” doktorlara yol gösterici bir harita sunar. Bu kriterler, Marfan sendromunun tanısını standart hale getirir ve hastalığın anlaşılmasını kolaylaştırır. [6] [7]
| Genetik bozukluk | İlgili genler/proteinler | Klinik özellikler |
|---|---|---|
| Loeys-Dietz sendromu (LDS) | TGFBR1/2 | Arteriyel tromboz, hiperlaktatemi, miyokardiyal sorunlar, arteriyel anevrizmalar, transnüüs anomalileri, tubuler sinyal defektleri, yüz displazisi, kafa derisi defektleri |
| Shprintzen-Goldberg sendromu (SGS) | FBN1 | Kraniyosinostoz, zihinsel gerilik |
| Ehlers-Danlos sendromu (EDS) – Artrokalazi tip (EZCA) | FBX2 | Eklem hiperlaksitesi, cilt frajilitesi |
| Marfanoid sendrom (WMS) | FBX1, ADAMTS10 | Mitokondriyal disfonksiyon, kardiyak defektler, eklem sertliği |
| Ectopia Lentis sendromu (ELS) | LTBP2, ADAMTS14, CBS | Aort anevrizması, lens dislokasyonu |
| Homosistinüri | – | Tromboz, zihinsel gerilik |
| Familial torasik aort anevrizması (FTAA) | TGFBR1/2, ACTA2 | Marfanoid habitus, livedo retikülaris, yüz anomalileri |
| Biküspid aort kapak (BAV) | – | – |
| Patent duktus arteriozus (PDA) ile FTAA | MYH11, SLC2A10 | Genel arteriyel tromboz, arteriyel stenoz, yüz deformiteleri |
| Arteriyel tortuozite sendromu (ATS) | – | – |
| Ehlers-Danlos sendromu (Vasküler tip) | COL3A1, COL1A2, PLOD1 | Büyük arter anevrizmaları, peritoni kolli, yarık damak/damak arkı, cilt transülensisi, düz saçlar, yüz çizgileri |
Marfan sendromunun tanısında en önemli ipuçları, “büyük bulgular” olarak adlandırılan belirgin özelliklerdir. Bunların başında vücudun ana atardamarı olan aortun genişlemesi gelir. Aortun duvarı zayıflayarak genişler ve bu durum özellikle aort kökünde görülür. Bu genişleme, hayatı tehdit eden bir yırtılma riski oluşturabilir. Doktorlar, ekokardiyografi veya MR gibi görüntüleme yöntemleriyle aortun genişliğini ölçerek bu riski değerlendirir.
Bir diğer önemli ipucu ise göz merceğinin yerinden kaymasıdır. Bu durum, Marfan sendromunun en karakteristik özelliklerinden biridir. Göz merceği normal pozisyonundan saparak hastaların bulanık veya çift görmesine neden olabilir. Özellikle genç yaşta ortaya çıkan bu belirti, Marfan sendromu tanısını güçlü bir şekilde destekler.
Aile öyküsü de tanı sürecinde kritik bir rol oynar. Marfan sendromu, ebeveynlerden çocuğa geçebilen kalıtsal bir hastalıktır. Bu nedenle, ailede Marfan sendromu öyküsü varsa, tanı koymak daha kolay hale gelir.
Marfan sendromu nedir sorusunu açıklarken teşhis için başvurulan büyük bulguların yanı sıra, “küçük bulgular” da tanıyı destekleyen önemli ipuçlarıdır. Örneğin, Marfan sendromlu bireyler genellikle yaşıtlarına göre daha uzundur ve kollar, bacaklar, el ve ayaklar orantısız şekilde uzun olabilir. Bu durum özellikle çocukluk çağında dikkat çekicidir. Ayrıca göğüs kafesinde içe çökme veya dışa çıkıntı gibi şekil bozuklukları da sık görülür. Omurga eğriliği (skolyoz) veya kamburluk gibi iskelet anomalileri de tanı sürecinde önemli bir yer tutar.


Tanıyı destekleyen bir diğer önemli araç ise genetik testlerdir. FBN1 genindeki mutasyonlar, Marfan sendromunun moleküler temelini oluşturur. Bu gen, vücuttaki bağ dokusunun yapısını destekleyen fibrillin-1 proteinini üretir. Bugüne kadar 500’den fazla FBN1 mutasyonu tanımlanmıştır. Genetik testler, özellikle klinik bulguların belirsiz olduğu durumlarda tanıyı netleştirmek için kullanılır.
Ancak Marfan sendromunun tanısı her zaman kolay değildir. Ki yazıda vurgulandığı üzere Marfan sendromu nedir sorusuna birden fazla cevap verilebilmesi de bu yüzdendir. Hastalık, diğer bağ dokusu bozukluklarıyla benzer belirtiler gösterebilir. Bu nedenle, ayırıcı tanı yapmak için dikkatli bir klinik değerlendirme ve genetik testler gereklidir. Özellikle TGF-β reseptör mutasyonları, Marfan benzeri sendromlara yol açabilir ve bu durum tanıyı daha da karmaşık hale getirebilir.
Erken tanı ile Marfan sendromu belirtileri teşhisi bu hastalığın yönetiminde hayati bir öneme sahiptir. Özellikle kalp-damar komplikasyonlarının önlenmesi açısından beta blokerler gibi ilaçlar aort genişlemesini yavaşlatabilir. Hatırlatmak gerekirse beta blokerler, birinci kalp krizinden sonra ikinci kalp krizini önleyici niteliktedir. Cerrahi müdahaleler ise aort yırtılması riskini azaltabilir. Ayrıca düzenli göz muayeneleri ve iskelet sistemi takibi, hastaların yaşam kalitesini artırmada önemli bir rol oynar.
Marfan sendromu tedavisi
Marfan sendromu nedir sorusunu cevaplandıktan hemen sonra elbette ki tedavi seçenekleri konuşulmalı. Peki hangisi için nasıl bir tedavi? Marfan sendromu, vücudun birçok sistemini etkileyebilen genetik bir bağ dokusu hastalığıdır. Tedavi süreci hastalığın karmaşık yapısı nedeniyle farklı uzmanlık alanlarından doktorların iş birliğini gerektirir. Kalp-damar sistemi, gözler, iskelet ve kas sistemi gibi birçok alanın düzenli takibi ve uygun tedavi yöntemleriyle hastaların yaşam kalitesi önemli ölçüde artırılabilir.
Kalp ve damar sistemi, Marfan sendromu tedavisi konusunda en çok dikkat edilmesi gereken alandır. Aortun genişlemesi (anevrizma) ve yırtılma riski, hastaların yaşamını tehdit edebilir. Bu nedenle kalp sağlığının korunması, tedavinin temelini oluşturur. Beta blokerler ve ACE inhibitörleri gibi ilaçlar, kalp atış hızını ve kan basıncını düşürerek aort üzerindeki baskıyı azaltır. Bu ilaçlar, aortun genişlemesini yavaşlatabilir ve yırtılma riskini minimize edebilir. Ancak aort genişlemesi kritik bir boyuta ulaştığında cerrahi müdahale gerekebilir. Aort onarımı veya değişimi gibi ameliyatlar, hayat kurtarıcı olabilir. Ayrıca mitral kapak problemleri de sık görülür ve bu durumlarda kapak tamiri veya protez kapak takılması gerekebilir. Marfan sendromlu bireylerin düzenli kalp taramalarıyla takip edilmesi kalp sağlığının korunmasında büyük önem taşır. [5]

Gözler de Marfan sendromundan önemli ölçüde etkilenir. Göz merceğinin yerinden kayması (lens çıkığı), bulanık veya çift görmeye neden olabilir. Bu durumda gözlük veya kontakt lenslerle görme düzeltilebilirken şiddetli vakalarda cerrahi müdahale gerekebilir. Retina dekolmanı gibi acil durumlar ise görme kaybına yol açabilir ve hemen tedavi edilmelidir. Ayrıca glokom ve katarakt gibi göz problemleri de erken yaşlarda ortaya çıkabilir. Bu nedenle Marfan sendromlu bireylerin yılda en az bir kez kapsamlı bir göz muayenesinden geçmesi önerilir.
İskelet ve kas sistemi, Marfan sendromu tedavisi konusunda bir diğer önemli alandır. Kardiyovasküler sistemden sonra en çok Marfan sendromu nedir sorusunun kaynağı olduğu için tedavisi de hayli merak konusu olmuştur. Uzun boy, uzun kollar ve bacaklar, omurga eğriliği (skolyoz) ve göğüs kafesi deformiteleri sık görülür. Bu durumlar, hastaların fiziksel görünümünü etkileyebilir ve hareket kabiliyetini kısıtlayabilir. Fizik tedavi, omurga esnekliğini ve kas gücünü artırmak için önemli bir rol oynar. Ortezler (destekleyici cihazlar), düz tabanlık veya skolyoz gibi durumlarda kullanılabilir. Şiddetli skolyoz vakalarında ise omurga düzeltme ameliyatı gerekebilir. Göğüs kafesi deformiteleri, solunum fonksiyonlarını da etkileyebilir ve bu durumlarda cerrahi müdahale gerekebilir. Kemik sağlığını korumak için D vitamini ve kalsiyum desteği de önerilebilir.
Solunum sistemi de Marfan sendromundan etkilenebilir. Akciğer çökmesi (spontan pnömotoraks); bağ dokusunun zayıflığı nedeniyle ortaya çıkabilen, ciddi bir durumdur. Ani göğüs ağrısı ve nefes darlığı gibi belirtilerle kendini gösteren bu durum, acil tıbbi müdahale gerektirir. Küçük vakalar kendiliğinden düzelebilirken ciddi durumlarda cerrahi müdahale gerekebilir. Ayrıca uyku apnesi gibi solunum problemleri de Marfan sendromlu bireylerde görülebilir. Bu durum, uyku sırasında solunumun durmasına neden olabilir ve CPAP cihazı kullanımı veya kilo kontrolü gibi marfan sendromu tedavisi için yöntem olarak değerlendirilebilir.
Marfan sendromu, deri ve bağ dokusunu da etkileyebilir. Ciltte çatlaklar (stria) sık görülür ancak bu çatlaklar genellikle zararsızdır. Kozmetik tedaviler -özellikle lazer uygulamaları- bu çatlakların görünümünü azaltmada etkili olabilir. Nemlendirici kremler de cilt elastikiyetini destekleyebilir.
Marfan sendromu tedavisi konuşulduğu zaman psikolojik ve sosyal destek, Marfan sendromlu bireyler için büyük önem taşır. Hastalığın fiziksel belirtileri, özellikle genç bireylerde beden algısını ve özgüveni olumsuz etkileyebilir. Psikolojik danışmanlık, bu tür duygusal zorluklarla başa çıkmada yardımcı olabilir. Ayrıca benzer durumdaki bireylerle deneyim paylaşımı yapabilecekleri destek grupları, hastaların motivasyonunu artırabilir. Tüm bunların yanında genetik danışmanlık, Marfan sendromu gibi kalıtsal bir hastalıkta aile planlaması yapmayı düşünen bireyler için büyük önem taşır. Prenatal tanı veya preimplantasyon genetik tanı (PGT) gibi seçenekler hastalığın gelecek nesillere aktarılma riskini azaltmada yardımcı olabilir. [8]
Marfan sendromu, erken teşhis ve düzenli takip ile yönetilebilen bir durumdur. Kardiyolog, göz doktoru, ortopedist ve genetik uzmanı gibi farklı alanlardan uzmanların iş birliği; hastaların yaşam kalitesini artırmada hayati bir rol oynar. Toplumda genetik hastalıklarla ilgili farkındalığın artırılması hem Marfan sendromlu bireyler hem de genetik yatkınlığı olan kişiler için daha iyi bir yaşam standardı sağlanmasına yani marfan sendromu tedavisi konusunda fazlasıyla yardımcı olabilir. Marfan sendromu nedir ifadesine de cevap bulabilmek tam olarak bu sebepten ötürü ziyadesiyle önemlidir. Gelecekte genetik araştırmaların ilerlemesiyle bu tür hastalıklar için daha etkili tedavi yöntemlerinin geliştirilmesi umut vericidir. Mevcut tedavi ve yaşam tarzı önerilerine uyum, hastaların sağlığını korumada en önemli adımdır.
- AKADEMİK DERGİ Collod-Béroud, G., & Boileau, C. (2002). Marfan syndrome in the third Millennium. European Journal of Human Genetics, 10(11), 673–681. [Makale Bağlantısı]
- AKADEMİK DERGİ Franken, R., Heesterbeek, T. J., De Waard, V., Zwinderman, A. H., Pals, G., Mulder, B. J., & Groenink, M. (2014). Diagnosis and genetics of Marfan syndrome. Expert Opinion on Orphan Drugs, 2(10), 1049–1062. [Makale Bağlantısı]
- AKADEMİK DERGİ Pepe, G., Giusti, B., Sticchi, E., Abbate, R., Gensini, G., & Nistri, S. (2016). Marfan syndrome: current perspectives. The Application of Clinical Genetics, 55. [Makale Bağlantısı]
- AKADEMİK DERGİ Cañadas, V., Vilacosta, I., Bruna, I., & Fuster, V. (2010). Marfan syndrome. Part 1: pathophysiology and diagnosis. Nature Reviews Cardiology, 7(5), 256–265. [Makale Bağlantısı]
- AKADEMİK DERGİ Erentuğ, V., Polat, A., Kirali, K., Akinci, E., & Yakut, C. (2005). Marfan sendromunda kardiyovasküler tutulum ve tedavi [Cardiovascular manifestations and treatment in Marfan syndrome]. Anadolu kardiyoloji dergisi : AKD = the Anatolian journal of cardiology, 5(1), 46–52. [Makale Bağlantısı]
- AKADEMİK DERGİ Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B., Hilhorst-Hofstee, Y., Jondeau, G., Faivre, L., Milewicz, D. M., Pyeritz, R. E., Sponseller, P. D., Wordsworth, P., & De Paepe, A. M. (n.d.). The revised Ghent nosology for the Marfan syndrome. Journal of Medical Genetics, 47(7), 476–485. [Makale Bağlantısı]
- AKADEMİK DERGİ Angel, M., Perez-Villardon, B., Vivancos-Delgado, R., & De, M. (2012). Marfan Syndrome – Advances in Diagnosis and management. In InTech eBooks. [Makale Bağlantısı]
- AKADEMİK DERGİ De Maio, F., Fichera, A., De Luna, V., Mancini, F., & Caterini, R. (2016). Orthopaedic aspects of Marfan Syndrome: The experience of a referral center for diagnosis of rare diseases. Advances in Orthopedics, 2016, 1–6. [Makale Bağlantısı]
APA 7: Özkahya, F. F. & Axology Journal. (2025, February 15). Marfan Sendromu Nedir? – Belirtileri ve Tedavisi. PerEXP Teamworks. [Makale Bağlantısı]
Dergiyi görüntüle!









