






Marfan syndrome is an inherited disorder of the body’s connective tissue. This condition leads to distinct Marfan syndrome symptoms across the body from the skeleton to the heart. Management often involves treatments like beta blockers for Marfan syndrome to protect the aorta and improve long-term health outcomes for patients.
What is Marfan syndrome and its primary causes?
Marfan syndrome is a complex disorder that impacts the body’s connective tissue. This tissue acts like the “glue” that holds your cells, organs, and tissues together. It provides strength and flexibility to structures such as bones, blood vessels, heart valves, and eyes. When this tissue is weaker than it should be, it can lead to problems throughout the body.
The root cause of this condition is a genetic mutation. Specifically, it involves a defect in the FBN1 gene. This gene provides the instructions for making a protein called fibrillin-1. Fibrillin-1 is a crucial component of microfibrils which are essential for forming elastic fibers within connective tissue. These fibers allow tissues to stretch and return to their original shape.
When the FBN1 gene is mutated, the body produces either a limited amount of fibrillin-1 or a defective version of the protein. This results in weakened connective tissue that stretches abnormally when placed under any stress. This underlying weakness is what leads to the wide range of health issues associated with Marfan syndrome.

The question of what is Marfan syndrome is best answered by understanding its genetic basis. It is an autosomal dominant disorder. This means an individual only needs to inherit one copy of the defective FBN1 gene from one parent to have the condition. About 75% of people with Marfan syndrome have a parent who also has it.
In the remaining 25% of cases, the condition results from a spontaneous new mutation in the FBN1 gene. This means neither parent has the disorder. The mutation occurred for the first time in either the egg or sperm cell. Once this new mutation occurs, the individual can then pass it on to their children.

The severity of Marfan syndrome can vary significantly even among members of the same family who share the same mutation. Some individuals experience mild effects while others face life-threatening complications. This variability makes personalized medical management and regular monitoring essential for anyone diagnosed with the condition.
Understanding the cause of Marfan syndrome is the first step toward managing it effectively. Knowledge of the genetic defect helps doctors anticipate potential problems and create a proactive treatment plan. This plan is designed to address issues before they become severe, significantly improving a person’s quality of life.
Marfan syndrome symptoms and its physical manifestations
The signs of this genetic disorder are diverse. The marfan syndrome symptoms can affect many different parts of the body. They may be noticeable at birth or may not appear until adolescence or young adulthood. The presentation of these signs can be very different from one person to another.
One of the most visible sets of marfan syndrome symptoms involves the skeletal system. People with the condition are often unusually tall and slender. They typically have long arms, legs, fingers, and toes. This specific feature of having long, thin fingers is known as arachnodactyly or “spider fingers.”

Chest and spine deformities are also common skeletal manifestations of marfan syndrome. The breastbone may sink inward (pectus excavatum) or protrude outward (pectus carinatum). Curvature of the spine, known as scoliosis, is another frequent finding. These skeletal issues can range from mild cosmetic concerns to more severe problems that affect breathing.
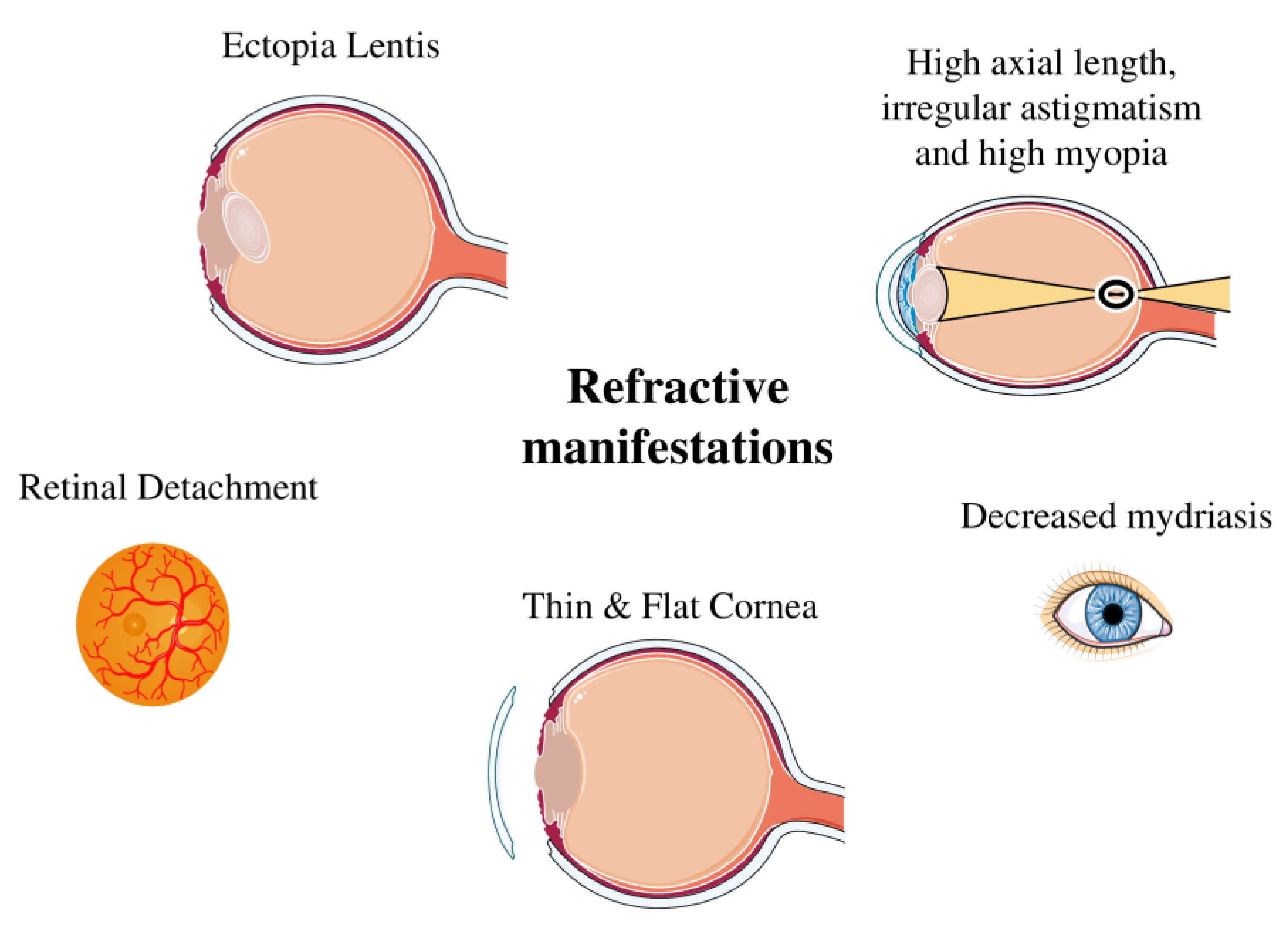
The eyes are another area significantly impacted. Over half of all individuals with Marfan syndrome experience a dislocation of one or both eye lenses. This condition, called ectopia lentis, is a hallmark sign. It occurs because the connective tissues holding the lens in place are too weak. Vision problems are therefore a major part of Marfan syndrome symptoms.
Other ocular issues include severe nearsightedness (myopia) due to an elongated eyeball shape. There is also an increased risk of developing early-onset glaucoma or cataracts. Furthermore, the weakened connective tissue in the eye puts individuals at a higher risk for retinal detachment, which is a serious medical emergency.
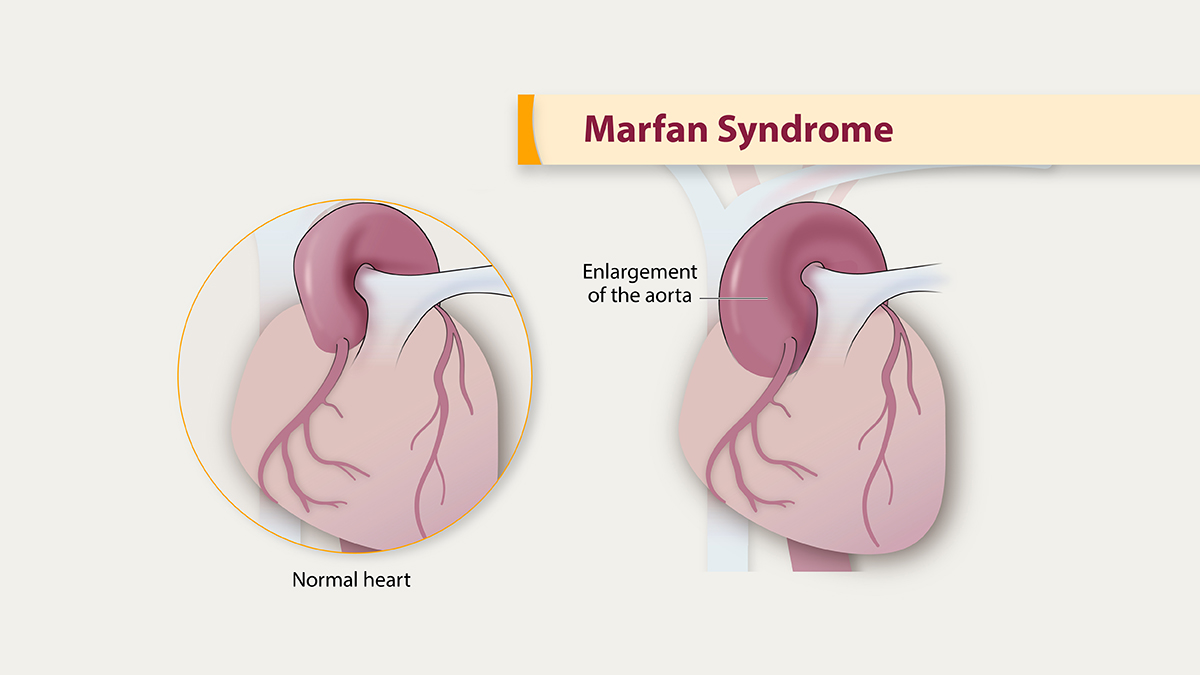
The most critical Marfan syndrome symptoms relate to the cardiovascular system, particularly the aorta. The aorta is the large artery that carries blood from the heart to the rest of the body. In Marfan syndrome, the wall of the aorta can become weak and stretch, a condition known as an aortic aneurysm.
This aneurysm can lead to a life-threatening aortic dissection where a tear occurs in the inner layer of the aorta’s wall. The force of the blood flowing through this tear splits the aortic wall, forcing the inner and middle layers apart from each other. This is a medical emergency that requires immediate intervention. Regular monitoring of the aorta is therefore a critical part of care.
The heart valves can also be affected. Mitral valve prolapse is common, where the valve between the heart’s left chambers doesn’t close properly. This can lead to blood leaking backward, causing a heart murmur, shortness of breath, or an irregular heartbeat. In severe cases, valve repair or replacement surgery may be necessary.
Other potential manifestations of marfan syndrome include effects on the skin and lungs. The skin may show stretch marks, known as striae, without any significant weight change. The lungs can also be affected, with an increased risk of a sudden collapsed lung (spontaneous pneumothorax) due to abnormal air-filled sacs in the lung tissue.
The role of beta blockers for marfan syndrome and other common medications
Managing Marfan syndrome focuses on preventing and treating its various complications. A key goal of medical therapy is to protect the aorta from enlargement and dissection. The primary treatment for this involves a class of drugs known as beta blockers. They are a cornerstone of modern care.
The use of beta blockers for Marfan syndrome is standard practice. These medications work by slowing down the heart rate and decreasing the force of each heartbeat. This reduces the pressure on the aortic wall, slowing the rate at which the aorta enlarges over time and lowering the risk of a dangerous tear.
Doctors typically prescribe beta blockers for Marfan syndrome as soon as a diagnosis is made, even in young children. The goal is to be proactive and protective. While they cannot reverse existing aortic damage, these drugs are highly effective at preventing further progression of the aneurysm, which is critical for long-term survival.
In addition to beta blockers, another class of blood pressure medications called angiotensin II receptor blockers (ARBs) is often used. Drugs like losartan have also been shown to slow aortic root enlargement. Sometimes, ARBs are prescribed alongside beta blockers for Marfan syndrome or as an alternative if a person cannot tolerate beta blockers.

These two drug classes form the primary line of defense for the cardiovascular system. The specific choice and dosage of Marfan syndrome medications are tailored to each individual’s needs. This decision is based on their aortic size, rate of enlargement, and overall health, requiring close collaboration with a cardiologist.
While cardiovascular health is the top priority, other medications may be used to manage different Marfan syndrome symptoms. For example, pain relievers may be necessary to address discomfort from skeletal issues like scoliosis or joint laxity. Eye drops might be prescribed to reduce pressure in the eyes and manage glaucoma.
Regular monitoring is just as important as medication. Individuals need frequent check-ups, including an annual echocardiogram to measure the aorta and check heart valve function. This consistent surveillance allows doctors to adjust treatment plans and intervene surgically if the aorta reaches a critical size, before a dissection occurs.
Surgical intervention is a vital part of management. If the aorta enlarges to a certain diameter (typically around 5 centimeters), doctors will recommend prophylactic surgery to repair or replace the aortic root. This preventative surgery has dramatically reduced the risk of fatal aortic dissection and is a major reason for improved life expectancy.
The success of treatment relies heavily on this combination of medical therapy, lifestyle adjustments, and regular monitoring. Taking beta blockers for Marfan syndrome every day, avoiding activities that strain the heart like competitive sports and heavy lifting, and attending all medical appointments are essential steps for managing the condition effectively.
What is the life expectancy with Marfan syndrome?
A diagnosis of a chronic genetic disorder naturally raises questions about the future. One of the most common concerns is about life expectancy. Fortunately, the outlook for individuals with Marfan syndrome has improved dramatically over the past few decades. It has transformed from a condition with a very poor prognosis to one that is highly manageable.
In the 1970s, before the benefits of proactive management were fully understood, the average life expectancy was only into the 30s or 40s. The primary cause of early death was aortic dissection. Today, with early diagnosis and modern medical and surgical treatments, the life expectancy for someone with Marfan syndrome approaches that of the general population, often reaching into the 70s and beyond.


This remarkable improvement is directly linked to better management strategies. The widespread use of medications like beta blockers for Marfan syndrome has been instrumental in protecting the aorta. These drugs, combined with regular imaging to monitor aortic size, allow doctors to intervene before a life-threatening event occurs.
Proactive aortic surgery has also been a game-changer. By repairing the aorta before it has a chance to dissect, surgeons can prevent the most dangerous complication of the disorder. Advances in surgical techniques have made these operations safer and more effective, contributing significantly to longer, healthier lives.
Living a full life with this condition requires a multidisciplinary team of healthcare professionals. This team often includes a cardiologist, an ophthalmologist, an orthopedist, and a geneticist. Coordinated care ensures that all potential Marfan syndrome symptoms are monitored and addressed promptly.

Genetic counseling is another important aspect of management. It provides individuals and families with information about the inheritance patterns of Marfan syndrome. This helps them make informed decisions about family planning and ensures that other family members who may be at risk can be tested and monitored if necessary.
While the medical advancements are significant, living with a chronic condition also involves emotional and psychological support. Support groups and counseling can help individuals and their families navigate the challenges of managing Marfan syndrome. A strong support network is invaluable for maintaining a positive outlook and adhering to a lifelong care plan.
Ultimately, the future for people with Marfan syndrome is brighter than ever before. Ongoing research continues to improve our understanding of the condition and develop new treatments. With diligent self-care, consistent medical follow-up, and a proactive approach, individuals can expect to live long and fulfilling lives.
- JOURNAL Judge, D. P., & Dietz, H. C. (2005). Marfan’s syndrome. The Lancet, 366(9501), 1965–1976. [Article Link]
- JOURNAL Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B., Hilhorst-Hofstee, Y., Jondeau, G., Faivre, L., Milewicz, D. M., Pyeritz, R. E., Sponseller, P. D., Wordsworth, P., & De Paepe, A. M. (2010). The revised Ghent nosology for the Marfan syndrome. Journal of Medical Genetics, 47(7), 476–485. [Article Link]
APA 7: TWs Editor. (2025, September 25). Marfan Syndrome: The Body’s Connective Tissue Disorder. PerEXP Teamworks. [Article Link]











